计算机药物设计绪论(Introduction to Computer-Assisted Drug Design)#
参考书目:
计算机辅助药物设计
Comprehensive Medicinal Chemistry II Volume 4
Computer-Assisted Drug Design
相关期刊:
Journal of Chemical Information and Modeling (JCIM)
Journal of Chemical Theory and Computation (JCTC)
Journal of Computational Chemistry (JCC)
Journal of Cheminformatics
PLoS Computational Biology (PLoS CB)
Briefings in Bioinformatics (BIB)
Bioinformatics
Nucleic Acids Research (NAR)
Nature Machine Intelligence
Journal of Medicinal Chemistry (JMC)
Journal of American Chemical Society (JACS)
Nature, Science, Nature Communications, etc
计算机辅助药物设计的发展史#
CADD技术诞生于药物研发的发展过程中。
药物研究与开发的历史,是个由粗到精,由盲目到自觉,由经验性的试验到科学的合理设计的过程,大致可以分为3个阶段:发现阶段(Discovery),发展阶段(Development),设计阶段(Design)。
药物研发的早期发现阶段:
偶然性大,缺乏目标
对毒副作用不够重视
对疾病的发病机制特别是分子层面的机制缺乏足够的认识
药物研发的发展阶段:
药物化学的发展阶段大致是在20世纪30年代到60年代。合成药物的大量涌现,是药物发展的“黄金时期”
分子药理学形成和酶学的发展,对阐明药物的作用原理起了重要的作用
药物研发的设计阶段:
一方面,爆发式增长过后,药物研发陷入低潮,系统性疾病的复杂性导致研究人员难以通过传统的方法获得有效的药物。研发经费激增,而成功率骤降
另一方面,“反应停”事件的出现导致了人们对于药物安全性的日益重视,也致使药物的研发难度大幅上升
反应停事件:1959年,西德各地出生过手脚异常的畸形婴儿。伦兹博士对这种怪胎进行了调查,于 1961年发表了“畸形的原因是催眠剂反应停”。反应停是治疗女性妊娠时早期孕吐的一种药物。
随着数据的积累,比如:靶标数目的增加、化合物实体库规模增长、复杂相互作用网络,常规实验手段不能满足指数增长的化合物的筛选和活性评价
药物设计的诞生得益于分子生物学、结构生物学、有机化学、物理化学、计算机科学等诸多学科的发展,其本身充分体现了学科交叉的特征
计算机辅助药物设计的概念#
计算机辅助药物设计(CADD)是以计算机作为操作媒介,利用计算化学、计算生物学、分子图形学、数理统计以及数据库等技术,研究药物和受体的相互作用,以期发现、设计和优化创新药物分子的方法学集合,其技术核心在于分子模拟,研究目的在于合理药物设计.
分子模拟是CADD的技术核心与实现手段
分子模拟是利用计算机以原子(量子)尺度模型来模拟分子的结构与行为,进而模拟分子体系的各种物理、化学性质的方法。它是在实验数据基础上,通过基本物理、化学原理,构筑的一套数理模型和算法。《中国大百科全书》
简单的说:计算理论和计算机图形学的结合
分子模拟的历史#
分子模拟的思想溯源于上世纪初量子力学的兴起:
1925年,Heisenberg发表了第一篇现代量子力学的文章
1926年,Schrodinger发表了著名的波动方程
1927年,采用量子力学的方法计算了氢分子的轨道,量子化学由此诞生
1930年,D. H. Andrews提出分子力学的基本思想
1933年,J. D. Bernal和R. H. Flowler提出了水的原子模型
1946年,Frank Westheiner完成了第一次分子力学计算
分子模拟的理论基础#
- 1964年,加州大学的沃尔特·科恩(Walter Kohn)教授和霍恩伯格(Hohenberg)博士从薛定谔方程出发,严格证明了一个重要性质:分子在基态的能量及其它所有性质可以由基态的电子密度的分布得到。即分子结构决定分子性质
Walter Kohn获得1998年诺贝尔化学奖
分子模拟的目的就是要用理论方法(模拟分子结构) 去实现以往用实验才能证明的东西(推导分子性质)
分子模拟的发展及意义#
1974年,Allinger等人发布了分子力场MM
1981年,Peter Kollman发布了AMBER力场
1983年,Martin Karplus发布了分子模拟程序CHARMM
…
分子模拟曾经限制在部分科学家手中,现在随着理论和计算机技术的发展,任何有兴趣的人都可以应用。而且应用范围越来越广,尤其在生命科学、药学、高分子、材料科学等多个领域取得了重要成果。21世纪也正好是这些领域大展身手的世界,分子模拟的作用也将更加重要。
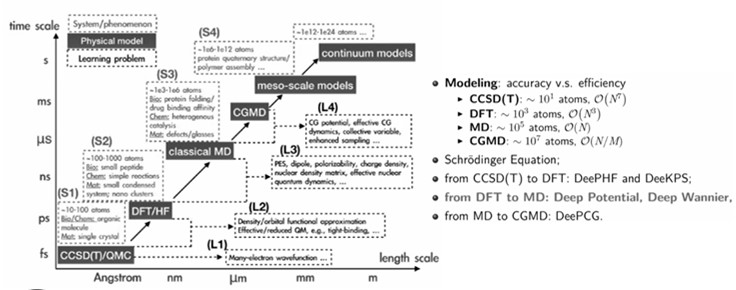
分子模拟的计算理论#
根据基本原理的不同,分子模拟可分为量子力学模拟和分子力学模拟
考虑电子运动状态 |
考虑核运动状态(电子运动作近似假设) |
||
量子力学模拟 |
用波函数表示 |
分子力学模拟 |
用质点运动轨迹表示 |
解薛定谔方程 |
解力学方程(经典牛顿力学方程) |
量子力学模拟(QM、QM/MM)
分子力学模拟
全原子模拟(all-atom simulation)
粗粒化模拟(coarse-grained simulation)
介观模拟(mesoscopic simulation)
连续介质模拟(continuum simulation)
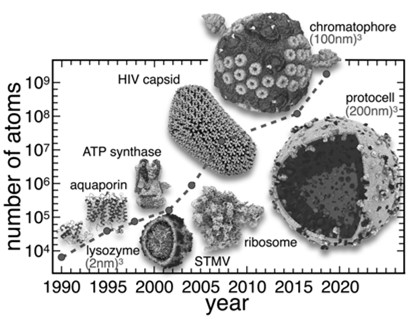
From the thousand-atom size scale, in the early 1990s, to a full protocell, on the billion-atom size scale,nowadays!!!
合理药物设计是CADD的研究目的#
合理药物设计(rational drug design)是指在分子病理学的基础上,依据靶点(target)的结构信息,发现和设计与之契合的药物分子,并充分考虑药代动及选择性等性质的药物设计过程。
合理药物设计包括:
基于结构的药物设计(sturucture-based drug design,SBDD)
基于性质的药物设计(property-based drug design,PBDD)
基于机理的药物设计(mechanism-based drug design,MBDD)
靶点(target)#
靶点主要指受体,广义上的受体包括酶、离子通道、膜蛋白受体、抗原-抗体、核酸等
基于结构的药物设计(SBDD)#
基于结构的药物设计包括:
基于受体结构的药物设计(target structure-based drug design, TSBDD)
直接药物设计方法
借助X射线晶体衍射、核磁共振或同源模建获得受体或受体-配体复合物的三维结构数据,经计算机图形学再现并设计药物分子
特点:目标清晰,设计准确,是首选的方法之一
基于配体结构的药物设计(ligand structure-based drug design, LSBDD)
间接药物设计方法
在受体三维结构不确定的情况下,从一系列已知活性分子出发(作用于同一靶标),分析其结构变化与生物活性强弱的关系,在计算机的辅助下,找出药效团,据此特征设计药物分子
特点:效率低,选择性差,结构新颖性不佳
计算机辅助药物设计的应用 The application of CADD#
CADD的应用范围#
在现代药物研发过程中,CADD技术已被广泛应用于各个环节:
靶点识别(bioinformatics)
生物靶标的结构特性表征
靶标可靶性评估
苗头化合物发现(hit)
药物-靶标相互作用特征分析
苗头分子结构优化与改造(hit to lead)
ADME/T性质的优化
临床分析(AI)
CADD的应用环境#
software: Algorithm and Logic
hardware: Computation and Visualization
分子模拟的常用软件
常用的综合软件平台:
Discovery Studio
Schrodinger
MOE
SYBYL
常用的开源软件:
Modeller,免费(https://salilab.org/modeller/)
Autodock,免费(https://www.autodock.scripps.edu/)
AMBER(Assisted Model Building with Energy Refinement)
GROMACS,免费(https://www.gromacs.org)
CHARMM(Chemistry at HARvard Macromolecular Mechanics)
CADD的技术组成:
分子力学(Molecular Mechanics,MM)
分子动力学(Molecular Dynamics,MD)
分子对接(Molecular Docking)
同源模建(Homology Modeling)
量化计算(Quantum Mechanical Calculation,QM)
定量构效关系(Quantitative Structure-Activity Relationships,QSAR)
药效团(Pharmacophore)
人工智能(Artificial Intelligence,AI)
数据库(Database)
分子力学(Molecular Mechanics):分子力学是分子模拟的基础之一,是连接微观机制与宏观表象的桥梁。
分子动力学(Molecular Dynamics):分子动力学是分子力学的应用拓展,其可进一步从时间尺度把握分子作用的微观机制。
分子对接(Molecular Docking):分子对接是分子力学的又一重要应用(基于力场的分子对接),其可快速识别可与目标靶点相结合的配体分子,是基于结构药物设计中最为重要的方法之一。
同源模建(Homology Modeling):同源模建通常以已知同源结构为模板,通过分子力学优化获得目标序列结构。
量化计算(QM Calculation):量化计算是分子模拟的又一重要分支,通过对电子波函数的精确描述,可有效把握化学反应的分子机制,是分子模拟中最为精确的理论研究方法。
定量构效关系(QSAR):定量构效关系(quantitative structure-activity relationships),是研究一组化合物的活性、毒性、药代性质与其结构(structural)、物理化学性质(physicochemical)、拓扑结构(topological)之间的相关关系,并用数理统计模型加以表征的研究方法。
药效团(Pharmacophore):药效团是产生特定药理作用所必须的物理化学特征及其在空间的分布,其是一组离散的物理化学特征在空间特定位置的分布,亦属3D-QSAR的一种。
人工智能(AI):基于人工智能的药物设计是近年最为热门的研究领域之一,其亦属广义QSAR模型。通过对已知数据建立复杂非线性映射关系来有效预测未知数据属性。
数据库(Database):数据库很多时候作为分子模拟数据来源,通过数据共享,极大节约时间、空间、资源/劳力成本,在CADD中发挥着极为基础的作用。
CADD中几个基础概念 Basic Concepts in CADD#
分子模拟中常用度量单位
距离单位:埃(Å,10^-10m)、纳米(nm,10^-9m)
角度单位:度(degree,°)、π(π = 180°)
时间单位:飞秒(fs,10^-15s)、皮秒(ps,10^-12s)、纳秒(ns,10^-9s)、微秒(µs,10^-6s )、毫秒(ms,10^-3s)
能量单位:kcal/mol、kJ/mol、hartree(a.u.)、eV(1 hartree = 27.2 eV = 627.5029 kcal, 1 kcal = 4.184 kJ)
温度单位:K(通常分子模拟温度为298.15 K或310 K)
坐标体系(Coordinate System)
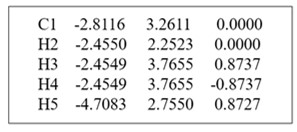
笛卡尔坐标(Cartesian Coordinates)即用原子的xyz值来表征分子构型。 如:甲烷分子的笛卡尔坐标表示法
内坐标(Internal Coordinates)用原子之间的距离、角度和二面角表征分子构型。通常用Z矩阵(Z-matrix)表示

两种坐标体系的比较:笛卡尔坐标一般用于定义含有大量原子的体系,如蛋白质、DNA等。对于一个含有n个原子的体系而言,其坐标数量为3N;笛卡尔坐标被广泛应用于分子力学模拟之中。分子内坐标一般用于定义含有较少原子的体系,如有机小分子化合物。对于一个含有n个原子的体系而言,其坐标数量为3N-6;分子内坐标一般应用于量子力学模拟之中。
常见的分子存储格式:
MDL公司的SDF(MOL)格式(无子结构信息、无原子类型、无部分电荷)
Tripos公司的MOL2格式(有子结构信息、有原子类型、有部分电荷)
蛋白晶体库的PDB(ENT)格式(有子结构信息、无原子类型、无部分电荷)
Accelrys公司的SMI格式(SMILES,仅字符串形式,无坐标相关信息)
目前使用的分子格式中,pdb格式广泛应用于生物大分子文件的存储(如蛋白质或核酸)
mol2格式常用于存储小分子信息,亦可用于生物大分子文件的存储(不常见)
其他形式的文件(sdf、mol、smi等)均用于小分子文件的存储(缺乏子结构定义形式)
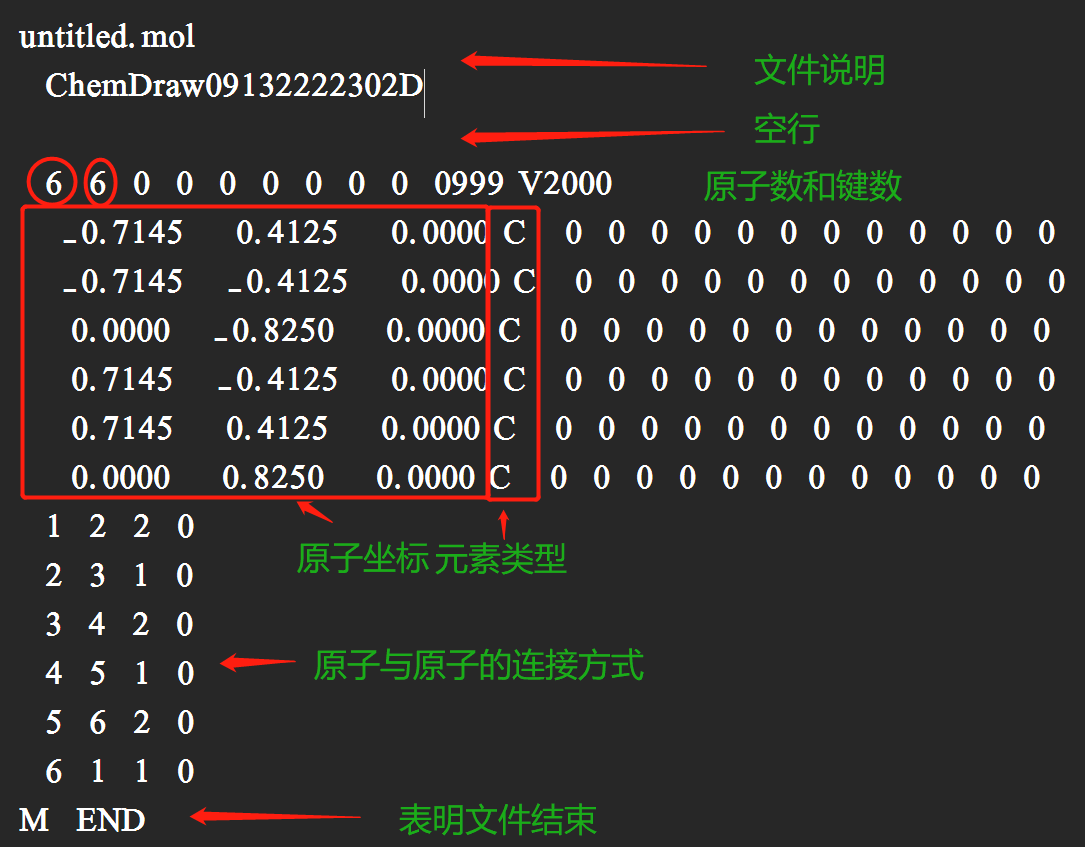
SDF(MOL)文件格式(苯分子):
MOL2文件格式(ALA氨基酸):

分子电荷形式:
形式电荷(formal charge):分子整体带电量(-1)
部分电荷(partial charge):又名原子电荷(atomic charge)
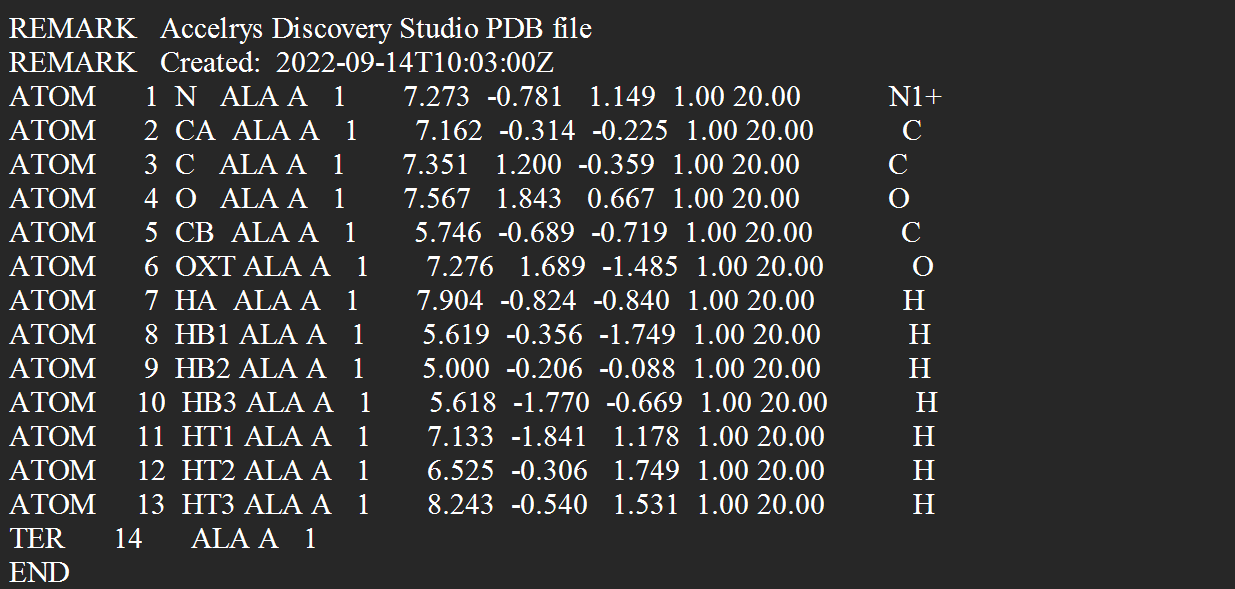
PDB(ENT)文件格式(ALA氨基酸):
SMI文件格式:分子线性输入规范(Simplified Molecular Input Line Entry System,SMILES)
计算效率极高,便于存储
使用传统化学符号(B, C, N, O, P, S, F, Cl, Br, I)
分子表述忽略H原子
CC |
ethane |
(CH3CH3) |
C=O |
formaldehyde |
(CH2O) |
C=C |
ethene |
(CH2=CH2) |
O=C=O |
carbon dioxide |
(CO2) |
COC |
dimethyl ether |
(CH3OCH3) |
C#N |
hydrogen cyanide |
(HCN) |
CCO |
ethanol |
(CH3CH2OH) |
[H][H] |
molecular hydrogen |
(H2) |
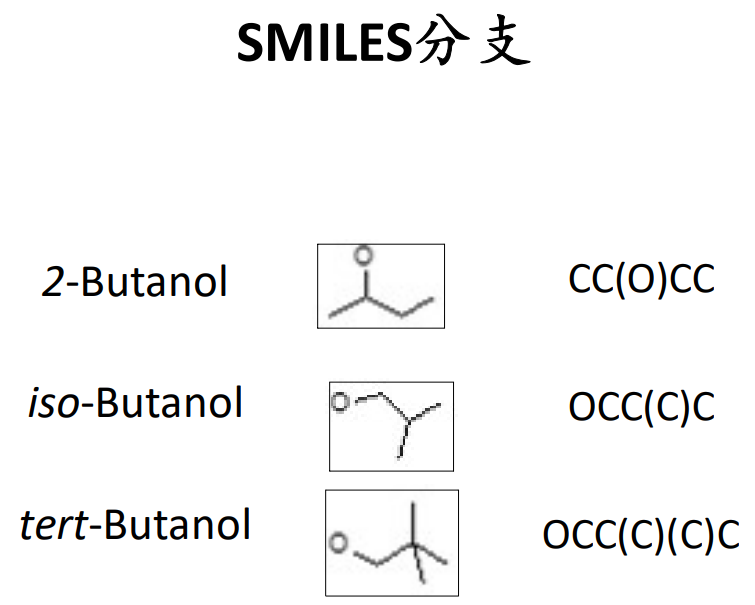
SMILES成环规则:
脂肪族或非芳香碳:C
芳香碳:c
成对数字表述闭合环结构 如c1ccccc1(或C1=CC=CC=C1)表示苯,而 C1CCCCC1表示环己烷
SMILES金属 [Al] [As] [Au] [Be] [Bi] [Cd] [Ca] [Fe] [Hg] [K] [Li] [Mg] [Na] [Ni] [Pt] [Sb] [Sn] [Zn] [Zr]
异构体与手性:
异构体原子使用“/”和“”,如:反式1,2-二氟乙烯:F/C=C/F 顺式1,2-二氟乙烯:F/C=CF
手性原子使用“@”
SMILES在人工智能模型中的应用:由于SMILES格式的文本格式,其在基于人工智能的分子生成模型中扮演着十分重要的角色
分子表面:
溶剂可及表面(solvent accessible surface):指探针分子在目标分子表面进行“假想滚动”时,其球心形成的轨迹(并非真实表面)。通常使用半径为1.4 Å的水分子作为探针分子。探针分子的中心可以放置于可及表面的任一点上,但不能穿入分子中任何原子的范德华球。
范德华表面(vdW surface):以原子的范德华半径生成的分子表面,由于缺少探针半径弥补,范德华表面通常更加离散
分子表面比较:
溶剂可及表面(solvent accessible surface)

范德华表面(vdW surface)

常用视图软件:
PYMOL
VMD