分子动力学模拟 (Molecular Dynamics Simulation)#
分子力学与分子动力学区别
分子力学方法:研究体系处于某一稳定状态的结构和性质,与“时间”无关
分子动力学模拟:研究原子和分子层面的动态行为,获得体系随时间变化的性质和规律,是真正的“计算机实验”
分子动力学基本原理 The principle of molecular dynamics simulation#
分子动力学里程碑#
1957年,Alder首次运用MD模拟,对刚性球体系进行研究(无势能函数,J. Chem. Phys. 1957, 27, 1208)
1974年,Rahman完成了第一个真正意义的MD模拟,即液态水模拟(J. Chem. Phys.1974, 60, 1545)
1977年,McCammon完成了第一个蛋白大分子的MD模拟(J. Andrew McCammon,Bruce R. Gelin & Martin Karplus, Nature, 1977, 267, 585–590)
分子动力学方法:利用牛顿力学基本原理,通过积分运动方程产生体系的连续构型,从而得到所有原子的运动轨迹,即体系中各粒子的位置和速度随时间变化的过程(坐标)
可通过对动力学轨迹的进一步分析获得体系的各种性质
牛顿运动定律:
物体总是连续地作匀速直线运动,除非一个外力作用于它
力等于动量与时间的比率,即 𝑓 = dp/dt = d(mv)/dt = m dv/dt = ma
对每一个作用都有一个大小相等方向相返的对抗作用
分子动力学一般步骤
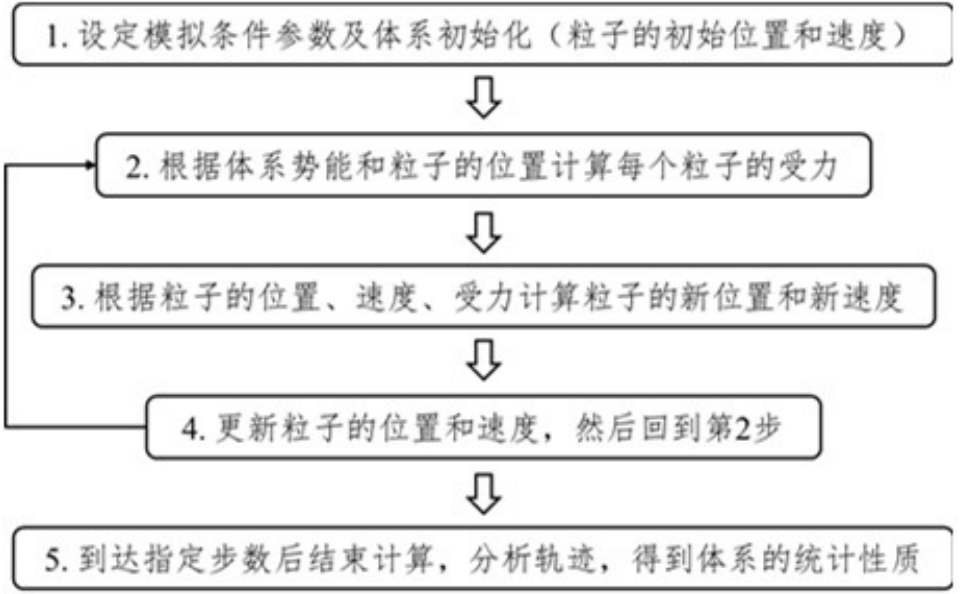
给定条件参数(温度、粒子数、模拟时间等)
体系初始化(初始位置和速度)
计算作用于所有粒子上的力
解牛顿运动方程,计算短时间内(time step)粒子的新位置
计算粒子新的速度和受力情况
重复3-5直至体系达到设置模拟时间。体系平衡后,等间隔保存原子的坐标,这些信息称为轨迹(trajectory)
分析轨迹数据,得到体系的统计性质
体系初始化:
初始化粒子的坐标(确立体系的初始构型):依据实验数据(X-Ray、NMR、冷冻电镜)、同源模建等
初始化粒子的速度(对每个原子指定初始速度):根据温度T时各粒子遵守Maxwell-Boltzmann速度分布随机生成
粒子受力求算:
按照经典力学理论,一个粒子受到的力等于体系势能函数对粒子坐标的一阶导数,其中势能由力场决定
在每个时间步长中,各原子的受力情况通过势能函数求算
两个原子之间的受力大小相等,方向相反
分子动力学中积分方法:
在一个连续势的作用下,所有质点运动被偶连在一起,引起一个多体问题,不能用解析法求解。在这种情况下,运动的方程只能利用有限差分方法来积分
有限差分方法的基本思想是把整体划分成许多小段,每一段通过固定的时间被分隔。在时刻t,体系中每个质点的总受力情况等于与其他质点相互作用的矢量加和
经过𝛿𝑡=(𝑡 − 𝑡_0)变化后,力𝐹_xi也发生改变
所以新的力𝐹_xi又被重新计算,从而又导出新的位置和新的速度
采用有限差值方法的分子动力学计算可以按照以下步骤进行:
模拟时间步长选择
在一个分子动力学模拟中,步长选择没有硬性规则
步长太小,有限时间内覆盖像空间不足
步长太大,不稳定性在积分算法中可能升高,由于原子间高能量的重叠,这种不稳定性可能导致模拟体系的崩溃
当模拟可柔分子时,一个有效的控制方式是时间步长应该设置为运动最快周期的十分之一的时间
最高频率振动是由于键伸缩,特别是与氢原子键合的键,一个C-H键振动具有一个近似10 fs的重复周期
因此一般时间步长应设置为1fs
一个真实的模拟通常会固定C-H键的伸缩,因此可以将模拟时间步长设置为2fs
边界条件(显示溶剂模拟)
在实际均一的溶液体系中,每个溶质分子(如蛋白质)周围都可以看作为一个无限的周期性环境
基本假设:
把溶质分子和周围的部分溶剂分子当作一个相对独立的单元(模拟体系)
如果研究体系受周围溶质或溶液的影响,则应采用周期性边界条件
当所关心的问题受周围环境影响较小时,模拟可以采用简单的非周期性边界条件
周期性边界条件:
周期性边界条件中基本单元形状的选择与模拟体系的形状有关:
最常用的是立方体和截顶八面体,采用这两种形状时,分子的考察最为简单。如果模拟的是一个核酸分子,则采用六方柱
如果模拟的是一个球形的蛋白质分子,则采用截顶八面体或菱形十二面体
非周期性边界条件:
当采用非周期性边界条件考虑溶剂效应时,在研究的重要部位外加入非周期的水分子环境,比如球形水环境
下图所示的水分子环境是以活性位点的重心为球心的半径为20Å的水分子球
在实际的分子力学或分子动力学模拟中,为了加快计算速度,可以把体系划分为柔性区(R1以内)、约束区(R1-R2之间)和完全刚性区(R2以外)

分子动力学轨迹分析#
分子动力学轨迹可以给出任何分子性质随时间的演化特征(含时性质),如体系构象转变过程、药物结合/解离过程等,常用于分析体系动力学性质的方法包括均方根位移(RMSD)、均方根振动(RMSF)、分子间相互作用能等
从单个轨迹点分析出的热力学性质为静态性质(特殊案例:分子力学优化能)
从多个轨迹点分析得出的热力学性质称为含时性质
能够预测含时性质是分子动力学最大优势之一
均方根位移 Root Mean Square Deviation(RMSD):计算体系在动力学模拟中随时间变化情况,可用于分析体系总体构象变化,判断其是否达到稳定状态
均方根振动 Root Mean Square Fluctuation(RMSF):计算动力学轨迹中每个片段(如氨基酸残基)的平均波动情况,可用于分析体系局部稳定性等情况
分子间相互作用计算
计算动力学轨迹中两片段相互作用情况(如药物-靶标结合自由能),由于考虑了分子间相互作用动态变化过程(含时性质),其计算结果比单点计算(如分子对接打分)更为严谨
分子力学优化及动力学模拟实验#
实验目的:#
掌握分子动力学模拟,观测体系构象变化。
熟悉 Discovery Studio 的基本操作。
熟悉 Protein Data Bank 数据库。
实验原理:#
使用 Discovery Studio 软件进行分子力学优化及动力学模拟。
- 本实验所用软件环境:
DS Version:19.1.0.18287 PP Version:19.1.0.1963 DS Client Version:19.1.0.18287 OS Distribution:Windows OS Version:10.0.22000
分子动力学操作流程 The process of MD simulation

分子初始构象获取:
小分子构象获取:
ChemDraw等软件画出结构
数据库获得
大分子结构获取:
PDB数据库获取
同源模建
分子预处理:
小分子结构预处理
分子状态重生成(异构体、质子化等)
大分子结构预处理
蛋白结构检测(去除非必须元素,主、侧链修复等)
质子化状态确定
蛋白质结构的预处理:通过实验方法所测得的生物大分子三维结构或多或少存在一些缺陷,比如分辨率低导致模型质量不佳、晶体结构无H原子和电荷信息、在模型构建步骤中相对粗糙的力场带来的结构误差等,这就需要在做SBDD前对生物大分子(如蛋白)的三维结构进行准备和能量最小化。
蛋白结构准备的一般原则:
缺失氨基酸及其侧链的修复
正确设置键级、计算电荷和加氢
限制性优化
水的处理
辅因子的处理
其它小分子的处理
Standard MD Cascade:
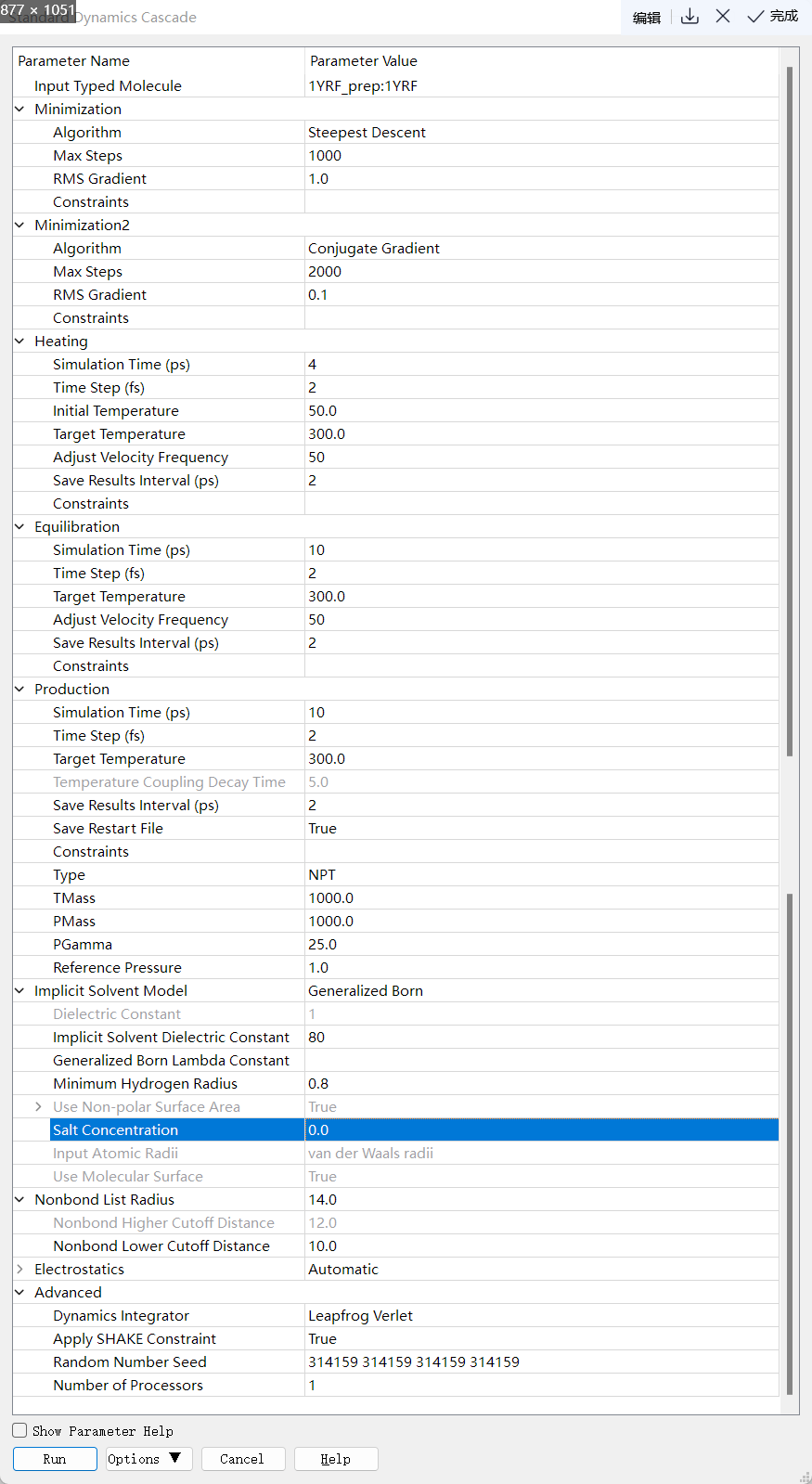
分子力学优化(Minimization):体系能量优化,结构优化的目的在于优化分子中因实验(低精度结构)或模建(加H或侧链修复等)产生的结构不合理问题,使用最陡下降法与共轭梯度法联合优化分子结构,以获得稳定初始状态。
升温期模拟(Heating):为了保持体系稳定,模拟初期需需从低温开始模拟(如50 K),逐渐升温至加室温(300 K)或体温(310 K)。体系首先根据Maxwell-Boltzmann分布随机生成初速度。升温过程需要逐步慢速进行,以防止体系不稳定。一般需要对体系进行一定限制。
平衡期模拟(Equilibration):升温过程结束后,即进入平衡期,平衡期模拟的主要作用是避免在加热过程中产生的局部或者全局的不稳定构象(T=300 K)。一般而言,经过平衡后,体系趋于稳定,可以开始后续的采样及分析。一般需要对体系进行一定限制。
产生期模拟(Production):产生模拟又称为采样模拟(sampling),是一个MD模拟有效数据的来源,也是MD模拟整个过程耗时最长阶段。此阶段通常不加任何限制。
数据分析:
动力学轨迹观察
RMSD计算
RMSF计算
实验步骤:#
获得分子起始构象:从 PDB 数据库网站(https://www.rcsb.org/)获取 HP53 蛋白晶体结构(PDB code: 1YRF)。(下载)
分子预处理:
(1)点击 Discovery Studio 软件上的 Macromolecules → Prepare Protein → Clean Protein 进行蛋白结构修复,矫正。
(2)点击 Discovery Studio 软件上的 Macromolecules → Prepare Protein → Prepare Protein 进行蛋白结构修复,矫正,质子化状态确定。设置参数如下:

Standard MD Cascade:点击 Discovery Studio 软件上的 Simulation → Run Simulation → Standard Dynamic Cascade 进行分子力学优化、升温期模拟、平衡期模拟、产生期模拟。设置参数如下:
结果分析 — RMSD、RMSF 计算:点击 Discovery Studio 软件上的 Simulation → Analyze Trajectory → Analyze Trajectory 进行 RMSD、RMSF 计算.设置参数如下:
