分子力学基本原理(Molecular Mechanics)#
分子力学的概念及发展(The concept and development of molecular mechanics)#
分子力学的起源:1930,D. H. Andrews
在分子内部,化学键都有“自然”的键长值和键角值。分子要调整它的几何形状(构象),以使其键长值和键角值尽可能接近自然值,同时也使非键作用(van der Waals)处于最小的状态,从而给出原子核置的最佳排布,即分子的平衡构型。
参考: Andrews, D.H. The relation between the Raman spectra and the structure of organic molecules, Phys. Rev. 1930, 36, 544
分子的经典力学模型:1946,T. L. Hill
Hill提出用van der Waals作用能和键长、键角的形变能来计算分子的能量,以优化分子的空间构型
Hill指出:“分子内部的空间作用是众所周知的,(1)基团或原子之间靠近时则会相互排斥;(2)为了减少这种作用,基团或原子会趋于相互离开,但是这将使键长伸长或键角发生弯曲,又会引起相应的能量升高。最后的构型将是这两种力折衷的结果,并且是能量最低的构型”
现代分子力场:1976, Norman L. Allinger
1976年,Allinger发布了MM1力场,1977年发布了MM2力场
虽然分子力学的思想和方法在19世纪30-40年代就已经建立,但是直到60-70年代以后,随着电子计算机的发展,用分子力学来确定和理解分子结构和性质的研究才越来越多。随着并行计算和超级计算机的普及,分子力学方法已经广泛应用于多个研究领域,如有机化学、生物化学、药物设计、材料设计等
分子力学(Molecular Mechanics)是使用基于原子核坐标的势函数和一系列相关势参数描述体系能量的方法
由于势函数和参数都是由经验和实验值拟合获得,故亦称经验力场方法(empirical force field method)
分子力学方法的建立和正确使用在于它的背景和若干假设
在分子模拟中许多问题涉及较大体系(如生物大分子、高分子体系等),无法用量子力学方法求解。其原因在于量子力学计算涉及体系的电子,以至于计算量远超计算机承受范围
分子力学方法忽略电子的运动,只计算以原子核的位置为函数的体系能量,因此可用于计算包含大量原子的体系 由于模拟参数的精确拟合,分子力场有时甚至可以获得近乎量子力学的精确解,而所需的模拟时间仅为量化计算的几十分之一甚至几百分之一,极大的提高了计算效率
注意:分子力学不能提供分子中依赖电子分布的性质
分子力学基本假设:
Born-Oppenheimer(波恩—奥本海默)近似假设(BO近似)
要点:核的振动运动比电子的运动慢得多,因此可近似地假设核的运动不影响电子的运动,他们是独立的
- 具体讲,∵M(质量)核>>M电子, V(速度)核<<V电子,
∴ 在解电子运动方程时可以近似地认为核的构型(位置)保持不变,即可把电子运动与核运动分开处理,核在电子的平均场中运动
显然,现实中不存在BO近似,把能量完全写做原子核坐标的函数并不准确
分子力学的模型表示形式:
分子模型假设:
球表示原子
弹簧表示化学健
弹簧的形变表示化学键的伸缩、弯折和扭转
非键结合的原子通过范德华和静电相互作用表示
分子力学基本特点:
分子力场不仅描述结构,也描述性质(结构决定性质)
一个完整的力场包括一套势函数以及相关的力场参数
不同的力场可以有相同的函数形式,但不同的参数
不同力场的参数(即使描述相同对象)不能混用
注意:力场都是经验的(相对从头计算而言),即没有绝对的正确与错误之分;分子力场参数都是拟合特定分子的数据而生成,因此只能说某个力场更适用于某些体系
计算的能量代表什么?
一个分子力学模型能简单地通过增加在所有键上的张力和所有原子间的范德华和库仑相互作用给出一个“能量”值。这个量称作E_MM,即分子力学能(也可称作空间(位)能)
E_MM最接近反映真实事物性质的是一个分子的内能:U标准的热力学方程,使U与其它量有如下关系:
对一个简单分子力学模型没有外部压力(真空中),有:
然而,自由能的变化(ΔG)与焓变(ΔH)和熵变(ΔS)有关,如下:
如果能使一个过程熵变足够小(ΔS ≈ 0),那么,
即:分子力学能量的变化ΔE_MM可以合理的近似等于自由能的变化ΔG
分子力场的组成形式 The composition of molecular force field#
分子力学的能量表达形式包括:
成键相互作用项
键伸缩能bond stretching/compression
键角弯折能angle bending
二面角扭转能torsion rotation
交叉作用项crossing terms
非键相互作用项
静电相互作用能electrostatic interactions
范德华相互作用能van der Waals interactions
附加项
以AMBER力场函数为例说明:

键伸缩能(Bond Stretching):Harmonic势函数
最基本的描述方法就是胡克定律(Hook’s law)

参考键长:当所有其它项在力场中值为零时,键长所采用的值
平衡键长:当体系处于能量最小时,键长所采用的值,此时其它项目对体系存在贡献
谐振子势函数在势井底部比较符合真实情况,即基态位置上比较符合实际,远离则不准确
键伸缩能(Bond Stretching):Morse势函数

AMBER 、CHARMM等采用谐振子函数形式
MM系列力场、MMFF94等采用莫斯函数
键角弯折能(Angle Bending):Harmonic势函数

谐振子模型在偏离平衡位置不大的情况下(10°以内)可以取得很好的结果
采用谐振子的力场包括:AMBER 、CHARMM等
二面角扭转能(Torsion Rotation)

注意:
由于二面角的扭转对总能量的贡献小于键长和键角的贡献,一般情况下二面角的改变要比键长和键角的变化自由得多
因此在一些处理大分子的力场中常保持键长、键角不变,只考虑二面角及其他的作用来优化整个分子的构象和能量。比如构象搜索就指定扭转角个数来寻找能量最低点
交叉相互作用项(Crossing Terms)

AMBER、CHARMM等力场中没有交叉相互作用项
MM2和MMFF94支持键伸缩-键角弯折相互作用项
静电相互作用(Electrostatic Interactions):由于不同元素吸引电子的能力不同,从而产生分子中电荷的不均衡(不规则)分布。描述这种电荷分布的方法有许多种。
电荷分布的表征和分布计算:
点电荷法
偶极矩法
点电荷法:

偶极矩法:根据一定规则计算出每个化学键的偶极矩,通过计算偶极-偶极相互作用来描述静电相互作用。

范德华相互作用(Van der Waals Interactions):
静电相互作用不能完全概括一个体系中所有非键相互作用。如在惰性气体原子中,多极矩为零,不存在偶极-偶极或偶极-诱导偶极相互作用,但实际存在原子间相互作用。这种偏差即是由范德华相互作用引起的
范德华相互作用是存在分子间的一种相互作用,主要有三种来源,即诱导力、色散力和取向力
范德华相互作用的计算:原子及分子间的色散吸引及交换排斥相互作用可通过量子力学来计算,这种计算通常考虑电子相关及采用较大基组,计算量巨大。在分子力学中通常使用较简便的方法计算,其中最有名的范德华势函数是Lennard- Jones 6-12函数(兰纳-琼斯势)
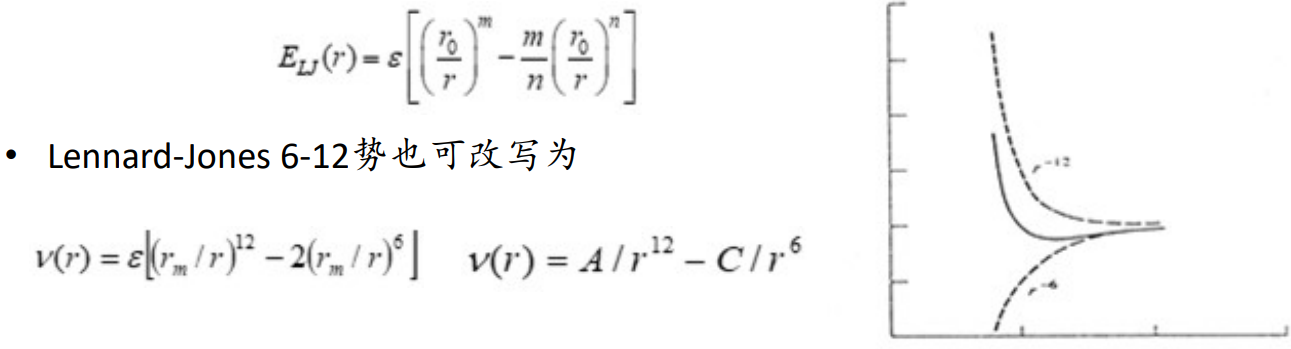
氢键作用(Hydrogen Bond)
H同时与两个原子相互作用
氢键能量在15~20 kJ/mol,而一般的共价键能量在400 kJ/mol
在生物体系中H一般与O、S和N形成氢键
由不同方法计算得到的能量绝对值无实际意义。只有当它与同体系的其它构象能量相比较时才有意义
比较不同程序计算得到的能量值无意义
用同一种程序时,比较不同分子的能量值无意义
力场的参数化
分子力场由两部分组:势函数形式,配套的力场参数
力场参数拟合的主要方法:实验拟合,量化计算拟合,此外,还有基于经验规则产生的力场参数(如Dreiding力场)
目标参数来源:
各类键长、键角的“本征值”一般取自晶体学、电子衍射或其他谱学数据
键伸缩和角变力常数主要由振动光谱数据确定
二面角扭转力常数通常由分子内旋转位垒(NMR谱带和弛豫时间、从头计算)来推算
非键相互作用参数则主要由晶格参数和液体的物理性质数据拟合得到
分子力场的种类 The type of molecular force field#
全原子力场(all-atom force field):考虑分子体系中的每一个原子,并对其分子参数进行精确定义。模拟最为精确,但计算量大
联合原子力场(combined force field):相对全原子力场进行了简化,忽略了体系中的非极性氢原子,将其参数整合到与它们成键的相邻原子上
粗粒化力场(coarse grained force field):进一步精简分子结构的力场参数,种类较多。例如,将氨基酸侧链看作一个颗粒而进行力场拟合。这一类力场通常针对特定体系
MMX力场
Allinger group, 1976
包括MM、MM2、MM3和MM4
主要适用于非极性有机小分子
函数形式比较复杂,包含交叉项
也可用于生物大分子体系,但是速度较慢
AMBER力场
Kollman group, 1981 * 最初仅为蛋白质和核酸体系提供相应的原子类型和力场参数 * 1990,发展了适用于多糖模拟的力场参数(Homan 1990) * 1995、2000、2004加入了适用于有机小分子的原子类型和参数 * 使用最广的分子力场之一(ff14SB+gaff) * 被SYBYL等综合商业软件所采纳
Wang, JM; Wolf, RM; Caldwell, JW; Kollman, PA; Case, DA, Development and testing of a general amber force field, J. Comput. Chem. 2004, 25, 1157. (citation>7500)
CHARMM力场
Karplus group, 1983
适用于各种分子性质的计算和模拟
对于从孤立的小分子到溶剂化的大生物体系都可以给出较好的结果
2009年加入CHARMM小分子力场使得应用范围更广泛(charmm36+CGenFF)
被Discovery Studio等综合商业软件所采纳
OPLS力场
Jorgensen group, 1985
将模拟液态小分子非键相互作用参数加入AMBER力场而产生的新力场
特别适用在液相系统中模拟体系物理性质
被不同分子模拟软件包广泛采纳,如GROMACS以及Schrödinger中的改良版
GROMOS力场
Gunsteren group, 1984
联合原子力场,力场参数主要以纯流体或混合流体体系在凝聚态下的热力学特征为实验数据拟合生成
适用于大分子体系的力场
计算速度较快
GROMACS软件原生力场
MMFF力场
Halgren group, 1996 (Merck Molecular Force Field)
基于MM3力场发展而来,定义了非常完备的原子类型 既适用于有机小分子,也适用于大分子体系,如蛋白质 被多个著名分子模拟软件包采纳,如Discovery Studio、MOE、SYBYL、RDKit
注意事项:
不同力场定义原子类型不同。因此,在选择力场时,需充分考虑所选力场能否完整表征目标体系中的所有原子
不同力场势能函数组成不同。力场对于不同的势能函数分项各有侧重,因此,应该充分考虑目标研究体系的主要特征,选择适应的力场。如有的力场考虑氢键,有氢键函数
不同力场势能函数参数不同。力场参数通过不同的实验体系拟合而成,如amber蛋白力场主要从蛋白大分子体系的实验数据中拟合参数,而 MM力场则从有机小分子体系中拟合参数注
常用分子力学优化算法 Molecular mechanics minimization#
分子力学为精确结构分析奠定了基础
众所周知,分子的物理、化学及生物学性质依赖于其三维结构(尤其是生物大分子中),即分子构象
虽然实验技术的发展使得分子结构测定变得普及,但是即使如此,仍然效率低、开销大。因此,分子稳定构象或优势构象的预测是分子模拟的重要任务之一
分子力学的发展为精确构象分析奠定了理论基础,其中能量优化算法为稳定分子构象的识别提供了算法保障分
能量优化的方法及特点
单纯形法(simplex algorithm):无需设置力场,通过调整原子位置排除位阻(如根据原子范德华半径判断位置是否 合适),精度较低,一般用于调整体系初始结构(如模建蛋白侧链调整)
导数法(derivative algorithm):需要设置力场,可提供势能面信息,导数的方向指向能量最小化的方向。可分为一阶导数法及二阶导数法
一阶导数方法
最陡下降法(Steepest Descent):最陡下降法是最常用的优化方法之一。能量梯度是进行搜索的方向,每次搜索之后旧的方向被新点处的梯度所取代。该方法对能量梯度依赖性较大,因此当体系能量远离最低点时,优化效率很高;但在最低点附近时,由于能量梯度接近于零,收敛很慢。适用于优化的初级阶段
共轭梯度法(Conjugate Gradient):通过当前的梯度和先前的最小化梯度比较确定能量最低点。在能量最低点附近时,收敛速度比最陡下降法快。
对于复杂体系(如蛋白体系),考虑到最陡下降法和共轭梯度法各有优势,因此实际应用中常采用二者联合的优化策略。如 在amber模拟中:首先以最陡下降法进行优化,以期快速降低体系总能量。随后采用共轭梯度法,加快在能量低点附近的收敛速率,获得能量最优构象。
二阶导数方法
牛顿-拉森法(Newton-Raphson):牛顿-拉普森法是一种二级微商算法,需要求解二阶导数矩阵(Hessian矩阵)。一级微商可确定优化方向,而二级微商可确定沿梯度在何处改变方向,因此这种算法的效率优于一阶导数法。由于在计算中需储存大量的二级微商,该算法计算量较大,速度慢,不适用于大体系。可首先以其他算法优化到能量最低点附近,再以此方法进行优化。
各优化方法的特点:
最陡下降法:方向变化大,收敛慢,优化幅度大
共轭梯度法:收敛快,易陷入局部最小值,对初始结构偏离不大
牛顿-拉森法:计算量大,当微商小时收敛快优化策略:可以多方法联合使用
注意事项:
结构优化往往只是局部优化,因此获得稳定构象只是初始结构附近的“最优构象”,而往往不是全局最优构象,因此结构优化的结果与初始构象的选择密切相关
若想找到全局最优构象,则需要将所有可能的初始构象进行优化,然后通过能量的比较获得全局最优构象(全局构象分析)
对于复杂分子体系,由于体系初始构象可能随自由度的增加而指数增加,因此往往很难通过简单优化策略获得全局最优构象
构象搜索与分析 Conformation searching and analysis#
构象分析简史
1950年前后,德里克·巴顿(Derek Barton)在取代环已烷反应性的研究中指出其反应性受取代分子的轴向性质影响,即受取代环已烷构象影响
1960年,亨得利克莱森(Hendricleson)用理论方法计算了甲基环己烷(methycyclohexane)扭船式与椅式构象转化过程的能量为6.7 kJ/mol,表明扭船式是可以存在的。直到1975年研究者才从实验中证实了环已烷扭船式的存在,表明基于计算的构象分析可以有效辅助实验观测。
事实上,甲基环己烷还有更多的局部最小构象(local minima)
构象分析方法(conformational search):
定义:指在构象空间中寻找所有可能出现的比较稳定的构象,即在分子的势能面上寻找所有极小值位点
目的:为了确定分子的优势构象
特点:不同于能量最小化,能量最小化的一个特征是只能优化到最接近初始结构的极小值位点;构象搜索可以搜索全空间(理论上)的极小值位点
能量势垒(Energy Barrier):当一个构象转变成另一个时,通常会遇到能量势垒,其可能来自于分子内部或分子间。势垒越高,结构转换越慢越难。
常用构象搜索算法 * 系统搜索法(仅用于小分子体系) * 片段连接法(常用于小分子体系) * 随机搜索法(常用于小分子体系) * 遗传算法(常用于小分子体系) * 模拟退火法(大分子、小分子体系均适用) * 分子动力学模拟方法(大分子、小分子体系均适用)
系统搜索法(格点搜索法):即在构象空间中以小的间隔变量进行逐点搜索,如果变量足够小,则可能搜索到全空间
步骤:
固定键长、键角
确定分子中可旋转键
设定步长
变化二面角,产生新构象并计算能量
案例:丙氨酸二肽势能面搜索

确定两个旋转角φ、ψ
固定键长键角
设定步长θi=300
计算体系能量(利用AMBER力场)
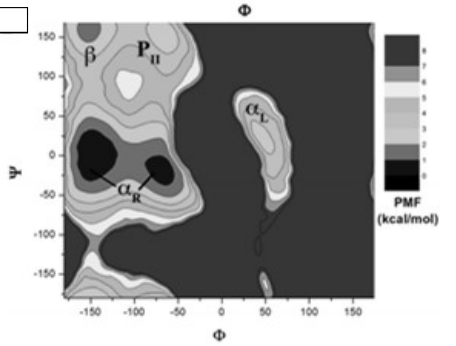
由图可见,两个区域特别重要,对应于α-螺旋(helix)和β-折叠(strand)结构
优越性:对于自由度较少的分子,系统搜索法得到的构象可以离散地覆盖整个势能面,即可以覆盖整个构象空间 缺点:计算效率低,只能应用于小分子。随着可自由旋转键的增加,构象数目呈指数增加。
θ_i是对键所选的两面角步长,N为旋转键的数
如N=5,θ=30 ,构象数为125,即248,832,69 hr(设1s计算一个构象)
如N=7,θ=30 ,构象数为127 ,即35,831,808 ,415天
片断连接法
系统搜索方法不能用于包含很多二面角的体系,在这个方法基础上发展起来的“片段连接(build-up)”方法可以在一定程度上缓解这个限制
基本思路:每种分子片段有其优势构象,通过把几个三维的“结构片断”相互连接组成一个完整分子来实现构象分析的过程
方法优势:这种方法比系统搜索更加有效的原因在于,与分子中可旋转单键相比,其组成“结构片断”的数目会少一些。这种方法尤其对于一些环系片断可能更为有效。常用于小分子体系
步骤:
确定构造整个分子需要哪些“结构片断”
产生所有片断的构象模式(储存于片断数据库中)
把“结构片断”相互连接
片断连接方法依赖条件:
结构片断的构象要相对独立。它的构象不依赖于与之连接的其他“结构片断”构象的变化
储存在数据库中的“结构片断”的构象模式应该覆盖这些“结构片断”在不同分子中所有可能的构象模式
内在缺陷:对于复杂分子,分子中基团之间的构象往往相互影响,“结构片断”的典型构型未必能够很好替代其在分子中的构象模式
随机搜索法:

改变笛卡尔坐标:识别分子中二面角,在每个原子的坐标x、y、z上随机加上随机量
改变内坐标:随机改变分子中可旋转键二面角
两种方法效率基本相等
随机搜索法的优势与缺陷:
比使用分子动力学的方法效率提高一个数量级
随机性较大,在有限搜索时间内,很难判断是否找到分子最佳构象
一般用于小分子体系
遗传算法
是一种借鉴生物界自然选择和自然进化的概率选择搜寻算法,具有高度并行、随机、自适应等特点
基本思路:以构象搜寻为例,个体由一系列可旋转键角度组成(染色体),通过大量个体的复制、变异、交叉互换产生新种群,并测量新种群(个体)的适应值(构象能量)来保留优势群体继续迭代搜索,最终得到优势构象
方法优势:并行度高,可同时获得一组优势构象

其中:
编码并产生初始化种群(产生大量分子构象)
计算个体适应值(如通过分子力学计算各构象能量)
复制、互换、变异产生新群体(不同个体的角度互换或随机更新)
遗传算法的特点与不足:
随机性较大,如初始种群的大小、突变概率、交叉概率的选取均可能导致十分不同的结果,参数的选择亦处于经验水平
对于自由度较多体系,所需初始种群的数量亦需指数增加,因此对于较大体系可能显著降低收敛效率,且计算量较大
常用于小分子体系
模拟退火法
也称作蒙特卡罗退火法,统计降温法或随机弛豫法。模拟退火方法是蒙特卡罗方法(Metropolis算法)搜集构象空间,其不同于传统蒙特卡罗方法之处在于运用体系能量的同时还把温度也作为体系的一个变量
整个过程分为两个步骤:
升温熔化体系
逐渐降温
在任一个温度下,体系的初始构象1,相应能量为E(1),构象发生微小的随机变化产生新的构象2,相应的能量为E(2),能量变化为ΔE=E(2)–E(1)
当ΔE<0时,接受构象变化; 当ΔE>0时,则在(0,1)间选择一个随机数R(如0.35),将其与P(ΔE)=exp(-ΔE/k_BT)相比较(Metropolis准则)若P(ΔE)>R则接受变化,新的构象成为下一次随机变化的起始点;否则拒绝变化,旧的构象仍是下一次随机变化的起始点。不断循环往复,在每个温度下都进行这个过程,直至体系温度降到足够低,即体系“冻结”在某个固定的构象上。
模拟退火法特点:
模拟退火效率如何取决于一些参数的设置,例如初始温度、降温因子以及随机数种子等,这些参数设定了退火的进程,决定了退火的效率
模拟退火方法的优点在于它取舍构象时不仅接受能量下降的变化,同时也接受部分能量上升的变化,因而有可能跳出局部势阱,寻找到新的能量最低点。同时模拟退火方法不依靠于体系起始构象,这样就消除了人为因素带来的影响
适用于大分子构象搜索
分子动力学方法
基本思想:根据分子的势函数,得到作用在每个原子上的力,利用牛顿第二定律求解运动方程,得到原子在势能面上的运动轨迹,从而达到构象搜索的目的
基于分子动力学模拟的构象搜索方法
淬火模拟(Quenching Simulation):高温模拟
退火模拟(Annealing Simulation):低温模拟
淬火模拟可以找到更多分子构象(高温易于翻越势垒),而退火模拟可以搜索出能量更低的构象
适用于大分子构象搜索